"Assessing the Ligand Properties of 1,3-Dimesitylbenzimidazol-2-ylidene in Ruthenium-Catalyzed Olefin Metathesis"
Yannick Borguet, Guillermo Zaragoza, Albert Demonceau and Lionel Delaude
 |
source: Dalton Transactions
year: 2013
volume: 42
first page: 7287
last page: 7296
doi: 10.1039/c2dt31520c
|
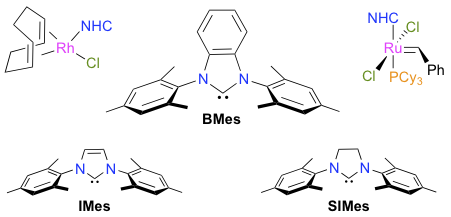
Abstract: The deprotonation of 1,3-dimesitylbenzimidazolium tetrafluoroborate with a strong base afforded 1,3 dimesitylbenzimidazol-2-ylidene (BMes), which was further reacted in situ with rhodium or ruthenium complexes to afford three new organometallic products. The compounds [RhCl(COD)(BMes)] (COD is 1,5-cyclooctadiene) and cis-[RhCl(CO)2(BMes)] were used to probe the steric and electronic parameters of BMes. Comparison of the percentage of buried volume (%VBur) and of the Tolman electronic parameter (TEP) of BMes with those determined previously for 1,3-dimesitylimidazol-2-ylidene (IMes) and 1,3-dimesitylimidazolin-2-ylidene (SIMes) revealed that the three N-heterocyclic carbenes (NHCs) had very similar profiles. Nonetheless, changes in the hydrocarbon backbone subtly affected the stereoelectronic properties of these ligands. Accordingly, the corresponding [RuCl2(PCy3)(NHC)(=CHPh)] complexes displayed different catalytic behaviors in the ring-closing metathesis (RCM) of α,ω-dienes. In the benchmark cyclization of diethyl 2,2-diallylmalonate, the new [RuCl2(PCy3)(BMes)(=CHPh)] compound (1d) performed slightly better than the Grubbs second-generation catalyst (1a), which was in turn significantly more active than the related [RuCl2(PCy3)(IMes)(=CHPh)] initiator (1b). For the formation of a model trisubstituted cycloolefin, complex 1d ranked in-between catalyst precursors 1a and 1b, whereas in the RCM of tetrasubstituted cycloalkenes it lost its catalytic efficiency much more rapidly.
[Full Text] [<< Previous Article] [Back to the List of Publications] [Next Article >>] l.delaude@ulg.ac.be